

Abstract: We present a rare case of disseminated histoplasmosis with haemophagocytic lymphohistiocytosis (HLH) in an immunocompetent individual. A 41-year-old female presented with a persistent fever, on and off for 2 months associated with weight loss, generalised weakness and shortness of breath on exertion. She had hepatosplenomegaly and pancytopaenia. After exclusion of routine causes of fever, a bone marrow evaluation was performed which was suggestive of histoplasmosis with features of haemophagocytosis. Urinary histoplasma antigen was positive. She was successfully treated with a combination of liposomal amphotericin B, intravenous immunoglobulins (IVIG), steroids and later itraconazole.
Key words: Histoplasmosis, Hemophagocytic Lymphohistiocytosis
Introduction
Histoplasma is a dimorphic fungus. Primary infection (histoplasmosis), is mostly benign, self-limiting and mild. Immunocompetent individuals rarely have widespread or disseminated infections. Our patient developed subacute infection with dissemination and had concurrent haemophagocytic lymphohistiocytosis (HLH). The requirement for immunosuppression along with antifungal treatment for disseminated fungal infection made this case a diagnostic and management challenge
Case report
A 41-year-old school-teacher from Delhi presented with complaints of low-grade intermittent fever and generalized weakness that had been progressively worsening over the past two months. Recently, she was unable to stand up from a sitting position without support. She also had a recent onset of dyspnoea on exertion even on walking <100meters on level ground. There was no associated orthopnoea paroxysmal nocturnal dyspnoea (PND). She had pain in the left submandibular region and pain in the right knee
Patient had travelled to Rishikesh 6 months prior to the presentation. She had no history of intake of alternative medications. Personal and family history was unremarkable
Clinical evaluation showed her to be febrile (990F) and dehydrated with no adventitious sounds on chest auscultation. She, however, had mild, tender hepato-splenomegaly. She also had tenderness in the left submandibular region as well as over the right knee. The initial laboratory investigations revealed pancytopaenia with haemoglobin (Hb) 9.8g/dL, total leukocyte count (TLC) 1.9x 109/L with absolute neutrophil count (ANC) 1370 X 109/L and platelet count 20 x 109/L. Patient had raised inflammatory markers with Ferritin 1080 ng/mL, C-reactive protein (CRP) 38 mg/L and erythrocyte sedimentation rate (ESR) 2mm. Lactate dehydrogenase (LDH) was 357 IU/L. Workup for infectious causes of fever was noncontributory. Autoimmune panel including antinuclear antibody-immunofluorescence (ANA-IF) and connective tissue disease profile was negative. Her angiotensin converting enzyme (ACE) levels were marginally raised and a possibility of sarcoidosis was considered. On imaging, ultrasound whole abdomen revealed hepatosplenomegaly and chest Xray revealed a fibrotic opacity in the right perihilar region (Figure 1).
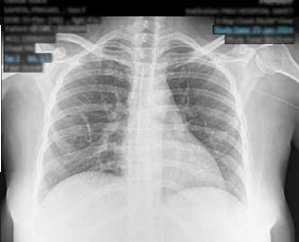
Figure 1: Chest X ray showing right perihilar opacity
Contrast-enhanced computer tomography (CECT) scan of chest and abdomen revealed fibro-atelectatic changes and mild hepatosplenomegaly. Echocardiography findings were mild pericardial effusion, no regional wall motion abnormalities (RWMA), left ventricle ejection fraction (LVEF) 60%, grade 2 diastolic dysfunction, mild pulmonary artery hypertension (PAH) (right ventricular systolic pressure [RVSP] 36 mmHg), no vegetations. A bone marrow evaluation was done and fungal organisms morphologically resembling Histoplasma capsulatum along with features of haemophagocytic lymphohistiocytosis were found. (Figures 2,3). Urine histoplasma antigen was positive. Viral markers for hepatitis B, hepatitis C and human
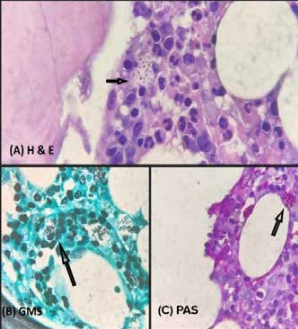
Figure 3: A, B and C – showing histoplasma species in bone marrow biopsy, stained by Gomori Methanamine Silver Stain (GMS) and Periodic Acid Schiff (PAS) (100x)
immunodeficiency virus (HIV) were negative. Fundus examination for ocular involvement was non-contributory.
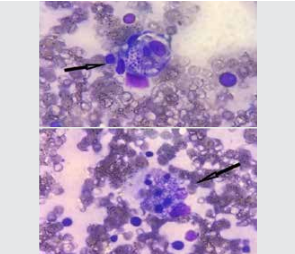
Figure 2: : Bone marrow aspiration showing haemophagocytosis with Histoplasma spp. (100x, MGG stain)
Management
With the establishment of the diagnosis, liposomal amphotericin B was initiated for treatment of histoplasmosis. The concurrent presence of HLH and its features necessitated the escalation of steroids. Intravenous immunoglobulin infusion (total of 100gms) was added in view of the worsening hyperferritinaemia. Filgrastim was given for significant neutropenia.
After 10 doses of amphotericin B, the patient was shifted to itraconazole 200mg twice daily. Therapy was planned for 1 year. On follow-up, the patient is afebrile, asymptomatic and pancytopaenia has resolved
Discussion
Histoplasma capsulatum is a dimorphic fungus commonly found in North and Central America, but its existence is documented in many diverse areas around the world. Most infected persons are either asymptomatic or have very few symptoms with mild illness.1 The various types of histoplamosis are pulmonary histoplasmosis, chronic cavitary pulmonary histoplasmosis, disseminated histoplasmosis and central nervous system infection. The diagnosis of the infection is done via antigen detection of the circulating H.capsulatum polysaccharide antigen in urine and serum.2 Our patient had a positive urine antigen test. Antibody tests are the complement fixation (CF) test utilizing two separate antigens, yeast and mycelial (or histoplasmin), and the immunodiffusion (ID) assay. A chemiluminescent DNA
probe for H. capsulatum is confirmatory. Direct visualization via histopathology can also be diagnostic as was seen in our patient on bone marrow examination.3
Haemophagocytic lymphohistiocytosis is a hyperinflammatory condition caused by excessive release of cytokines.4 There are eight diagnostic criteria of which five must be present for the diagnosis: (1) fever, (2) splenomegaly,(3) cytopenia in at least two lines with haemoglobin <9 g/dL, neutrophil count less than 100*1099 /L and platelet count less than 1*1099/L (4) hyperferritinaemia >500 μg/L , (5) hypofibrogenaemia <1.5 g/L or hypertriglyceridaemia >3mmol/L, (6) high soluble cluster of differentiation (CD)25 >2400U/ml, (7) haemophagocytosis in bone marrow, spleen, or lymph nodes, and (8) low or absent natural killer (NK) cell activity.5 This patient had fever, splenomegaly, pancytopenia, hyperferritinaemia and haemophagocytosis on bone marrow exam thereby meeting the diagnostic criteria
The current treatment of HLH is based on the HLH-2004 protocols. Majority of the patients can be managed with immune modifying treatment with either only intravenous steroid pulses (1 g of methylprednisolone for 1-3 consecutive days) followed by oral 1 mg/kg/d of prednisolone with appropriate rapid tapering of 10 mg every fourth day.5 In infection-triggered HLH, a high dose of IVIG should be considered in combination with steroids, along with antimicrobial agents which should also be concurrently started at the time of presentation. Anakinra can also be used.
Our patient was diagnosed as disseminated histoplasmosis with HLH. She was treated with a combination of lipid formulation of amphotericin B at a dosage of 3 to 5 mg/kg daily for 10 days which was then overlapped with oral itraconazole 200 mg twice daily which was continued as monotherapy twice daily, after clinical improvement. The HLH component was managed with methylprednisolone and later IVIG.
CONCLUSION:
Our patient was a diagnostic challenge. The prolonged fever with paucity of findings on physical examination as well as on laboratory investigations made the diagnosis obscure. The raised ACE levels lead her to be treated as a case of sarcoidosis which is one of the differential diagnoses of histoplasmosis. The clear evidence provided by the bone marrow evaluation clinched the diagnosis.
The second conundrum was the management of disseminated histoplasmosis with HLH which needed immunosuppression. Initially, steroids were escalated, then subsequent immunosuppression was achieved with IVIG infusion, with antifungal cover. Complications like hypokalaemia were managed conservatively.
A search of Pubmed with the keywords of histoplasmosis and haemophagocytic lymphohistiocytosis (HLH) brought up very few search results with ‘immunocompromise’ as a major factor in these case reports. Our patient is unique in being immunocompetent but nevertheless developing severe disseminated histoplasmosis and developing HLH along with it.
Jitesh K Sisodiya, Monica Mahajan, Vaibhav Rohatgi, Nitin Dayal, Pranjali Gupta, Shweta R Munot. Disseminated Histoplasmosis with Haemophagocytic Lymphohistiocytosis in an Immunocompetent Patient – A Rare Complication. MMJ. 2024, Sept. Vol 1 (3).
References
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45(7):807-25.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20(1):115-32.
- Hage CA, Wheat LJ. Histoplasmosis, Harrison's Principles of Internal Medicine, 21e Loscalzo J, Fauci A, Kasper D, et al (Eds.),Eds. Joseph Loscalzo, et al, pg 1658-1660.
- Koumadoraki E, Madouros N, Sharif S, et al. Hemophagocytic Lymphohistiocytosis and Infection: A Literature Review. Cureus. 2022;14(2):e22411..
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014 Apr 26;383(9927):1503-1516. Epub 2013 Nov 27. Erratum in: Lancet. 2014 Apr 26;383(9927):1464.