

Abstract:
Polyarteritis nodosa (PAN) is a rare, necrotising medium-vessel vasculitis characterised by multisystem involvement and the absence of anti-neutrophil cytoplasmic antibodies (ANCA), with an estimated prevalence of 3–4 cases per million annually. Early recognition is critical to prevent life-threatening vascular complications. A 68-year-old male with known coronary artery disease presented with one week of high-grade fever, dry cough, dyspnoea on exertion, lower-extremity oedema, transient chest rash, anorexia, and weight loss. Initial laboratory evaluation revealed leukocytosis, markedly elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), hyponatraemia, mild renal and hepatic dysfunction, and elevated N-terminal pro b-type natriuretic peptide (NT-proBNP) and ferritin. Imaging showed hepatosplenomegaly and perinephric fat stranding. Despite broad-spectrum antibiotics and corticosteroids, inflammatory markers remained elevated. Autoimmune serology was antinuclear antibody (ANA)-negative but positive for myositis antibodies (Mi-2, Ku, Ro-60) with low complement levels; ANCA were negative. On hospital Day 10, the patient developed sudden left flank pain, hypotension, lactic acidosis, and worsening renal function. Computed Tomography (CT) angiography demonstrated multiple microaneurysms in renal, hepatic, splenic, and mesenteric vessels with active bleeding from a left renal artery aneurysm and a large perinephric haematoma. Emergent coil embolisation and drainage were performed. Skin biopsy confirmed transmural fibrinoid necrotising vasculitis; nerve conduction studies revealed sensorimotor neuropathy. A diagnosis of PAN was established. The patient received high-dose intravenous (IV) methylprednisolone followed by oral steroids and azathioprine. Clinical and laboratory parameters improved rapidly, and he was discharged on maintenance immunosuppression in stable condition. This case is unique for its initial presentation mimicking sepsis and heart failure, the absence of ANCA, and life-threatening rupture of a renal artery aneurysm—an uncommon but recognised complication of PAN. In patients with fever of unknown origin and multisystem involvement unresponsive to antimicrobial therapy, medium-vessel vasculitis must be considered. Prompt angiographic evaluation and histopathologic confirmation facilitate early immunosuppressive treatment, reducing morbidity and improving survival in PAN.
Key words: Polyarteritis Nodosa, Fever, Vasculitis, Renal Artery Aneurysm, Perinephric Haematoma.
Introduction
Polyarteritis nodosa (PAN) is a rare necrotising medium-vessel vasculitis which affects multiple organs such as the nerves, kidneys, and gastrointestinal system. Data for PAN in India are limited. Initial case reports from the All India Institute of Medical Sciences (AIIMS), New Delhi, and the Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh described a range of clinical features, including constitutional symptoms, mononeuritis multiplex, hypertension, and gastrointestinal complications. Mononeuritis multiplex has persistently been established as the most common neurological manifestation, found in up to 83% of Indian patients.1 Diagnosis of PAN may be difficult because of its nonspecific clinical presentation and the lack of characteristic serological markers like antineutrophil cytoplasmic antibodies (ANCA). A negative skin biopsy is not conclusive for excluding PAN.2 Spontaneous perirenal haematoma due to ruptured microaneurysm is a rare but dangerous complication of PAN.3 In this article, we report the case of an elderly man who had spontaneous perirenal haematoma and was later diagnosed to have PAN.
Case Report
A 68-year-old male with a history of coronary artery disease (CAD) presented with high-grade fever, cough with minimal whitish sputum, exertional dyspnoea, bilateral pedal oedema, along with erythematous rash over the chest for a duration of one week. Over the last one month, he had a loss of appetite and weight.
here was no history of joint pain, oral ulcers, photosensitivity, or hair loss. He had no prior diagnosis of autoimmune disease or hepatitis. On examination, he was conscious and oriented with bilateral episcleritis, grade 2 pedal oedema. His blood pressure was 110/76 mmHg and pulse was 90/min. Systemic examination revealed bilateral infrascapular fine crepitations with decreased breath sound intensity, mild hepatosplenomegaly, and mild left flank tenderness. There was no lymphadenopathy, or focal neurological deficit noted.
Initial investigations are mentioned in Table 1.
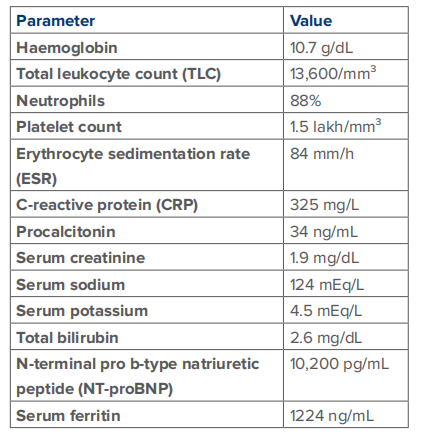
Table 1: Initial laboratory investigations.
Urinalysis was normal
Regular fever workup was negative for infectious disease aetiology, including blood and urine cultures. Chest X-ray showed bilateral pneumonitis. Computed tomography (CT) of the chest revealed interlobular septal thickening, pleural effusions, and cardiomegaly. CT of the abdomen showed bilateral perinephric stranding, hepatosplenomegaly, and cholelithiasis without cholecystitis.
Empiric antibiotics and steroids temporarily suppressed fever, but inflammatory markers remained elevated. Autoimmune screening revealed antinuclear antibody (ANA) immunofluorescence negative but ANA line immunoassay (ANA-LIA) panel positive for Mi-2, Ku, and Ro-60. Complement component 3 (C3) and complement component 4 (C4) levels were decreased. Cytoplasmic ANCA (C-ANCA) was indeterminate and perinuclear ANCA (P-ANCA) was negative.
On Day 10, the patient presented with acute-onset abdominal pain with hypotension (blood pressure 60/40 mmHg), elevated lactate (4.7 mmol/L), and deteriorating renal parameters.
CT angiography was performed and revealed multiple small and medium-sized artery aneurysms of both kidneys, hepatic and splenic branches, and the superior mesenteric artery, as well as an actively bleeding left upper pole artery aneurysm causing a large perinephric haematoma (Figure 1).
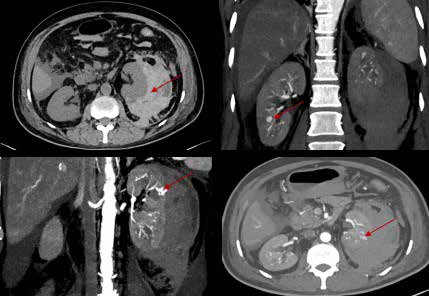
Figure 1: A: Non-Contrast computed tomography (CT) image showing perinephric collection of blood around the left kidney; B: Contrast-enhanced CT image of the right kidney showing aneurysm; C and D: Contrast-enhanced CT images showing "culprit" bleeding aneurysm.
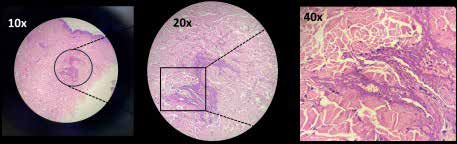
Figure 2: Fibrinoid necrosis of the vessel wall with perivascular inflammation.
Nerve conduction studies showed mixed axonal and demyelinating neuropathy in all four limbs. Hepatitis B surface antigen (HBsAg) and anti-hepatitis C virus (anti-HCV) antibodies were negative. The patient showed clinical improvement. Renal function and laboratory parameters stabilised, and he was discharged on azathioprine and low-dose steroids. He remained stable on follow-up with no recurrence of fever or abdominal symptoms.
Emergency angioembolisation of the left renal artery was performed, and a pigtail catheter was placed to drain the haematoma.
A diagnosis of PAN was made. He was treated with intravenous (IV) methylprednisolone pulse doses.
A skin biopsy from an erythematous rash over the abdomen revealed fibrinoid necrosis of the vessel wall with perivascular inflammation, consistent with vasculitis as shown in Figure 2.
Discussion
PAN, initially described by Kussmaul and Meier, was characterised by the 1994 Chapel Hill Consensus Conference (CHCC) as a necrotising arteritis of medium-and small-sized arteries, without glomerulonephritis.
The 2012 CHCC revision additionally defined PAN as an ANCA-negative vasculitis and introduced new categories, such as single-organ vasculitis and hepatitis B virus (HBV)-associated vasculitis, while distinguishing it from idiopathic PAN. To improve diagnostic consistency, the European Medicines Agency (EMA) subsequently developed a stepwise classification algorithm based on CHCC definitions and the American College of Rheumatology (ACR) criteria.4 A multicentric study by Joshi and Mittal, conducted across major Indian cities, compiled 1064 cases of vasculitis, of which 8.83% were PAN and 1.22% were cutaneous PAN. Conversely, Henoch–Schonlein purpura (21.8%), Takayasu arteritis (20.2%), and Wegener's granulomatosis (13.8%) were most commonly reported vasculitis types. Microscopic polyangiitis (MPA), which is easily mistaken for PAN, was seen in 3.94% cases. The results reflect the relative infrequency of PAN in the Indian population.1
Diagnosis of PAN remains challenging due to its protean clinical manifestations and the absence of a definitive diagnostic test. It can present with nonspecific manifestations such as fever, weight loss, myalgia, or neuropathy, and can present mimicking infections, malignancies, or other autoimmune conditions. Unlike other vasculitides, PAN tends to be ANCA-negative and typically does not involve arterioles, capillaries, or glomeruli, resulting in delayed diagnosis. The diagnosis of PAN in our patient was supported by the ACR 1990 classification criteria. These criteria, derived from a comparison of 118 PAN cases and 689 controls with other vasculitides, include ten clinical, laboratory, radiologic, and histopathologic features. The presence of at least three of the following—weight loss ≥4 kg, livedo reticularis, testicular pain, myalgias, mononeuropathy or polyneuropathy, diastolic blood pressure >90 mmHg, elevated serum creatinine or blood urea nitrogen, HBsAg positivity, angiographic abnormalities, and biopsy-proven arterial inflammation—confers a sensitivity of 82.2% and specificity of 86.6% for PAN classification.5
In our case, the diagnosis of PAN was initially obscured by overlapping features of sepsis with heart failure. The occurrence of a spontaneous perinephric haematoma from aneurysmal rupture, multisystemic involvement of peripheral neuropathy, acute kidney injury, episcleritis, and aggressive clinical course despite empiric antibiotics, all pointed in the direction of systemic vasculitis. This uncommon but characteristic angiographic feature, along with skin biopsy showing leukocytoclastic vasculitis, established the diagnosis of PAN. This case emphasises the need for diagnosing PAN in patients with fever of unknown origin and progressive multisystem disease, particularly with poor clinical response to infection-directed therapy. It also highlights the need to integrate clinical findings, radiologic evidence and histopathology rather than relying solely on serologic markers or autoantibody panels, which are potentially non-specific or misleading in ANCA-negative vasculitides such as PAN.6
The differential diagnosis of PAN comprises infections as well as other vasculitides. The most important infectious mimics are infective endocarditis, human immunodeficiency virus (HIV)-associated vasculitis, and mycotic aneurysms. The non-infectious mimics are ANCA-associated vasculitides—granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA)—as well as immunoglobulin A (IgA) vasculitis, cryoglobulinaemic vasculitis, and drug-induced vasculitis. Key distinguishing features include ANCA status, organ involvement, and histology.7
In patients with PAN linked to HBV or hepatitis C virus (HCV) infection, the primary emphasis of therapy is the administration of antiviral drugs for treatment of the underlying viral disease.8 Single-organ PAN is typically monophasic and non-recurrent. Isolated single-organ PAN is generally diagnosed histologically from surgical specimens, and treatment beyond surgical excision is typically not required.8 Mild cutaneous PAN, presenting with subcutaneous nodules and livedo reticularis, may respond to nonsteroidal anti-inflammatory drugs (NSAIDs), rest, and elevation of limb(s).9 In cases of severe active PAN, usual therapy consists of high-dose glucocorticoids combined with cyclophosphamide. IV methylprednisolone is generally administered in the dose range of 7–15 mg/kg (maximum of 1 g/day) over three consecutive days, followed by oral prednisone at 1 mg/kg/day for two to four weeks with tapering over time according to response.10 Cyclophosphamide may be given orally at 2 mg/kg/day or intravenously at 15 mg/kg at Weeks 0, 2, and 4, and subsequently every three weeks until remission.11 Following cyclophosphamide induction, patients are switched to a maintenance immunosuppressive drug like azathioprine (2 mg/kg/day) or methotrexate (20-25 mg weekly), with the overall duration of immunosuppressive therapy—induction and maintenance—usually lasting to 18 months. Relapsing patients can be treated with a more extended course.12 In our patient, cyclophosphamide was deferred because of ongoing haematuria and a recent vascular intervention. Immunosuppression was initiated with IV glucocorticoids, followed by azathioprine once appropriate thiopurine methyltransferase (TPMT) activity was confirmed.
With prompt and appropriate immunosuppressive therapy, the five-year survival in PAN can be greater than 80%.13 However, delays in diagnosis or therapy can result in catastrophic complications, including life-threatening phenomena like spontaneous aneurysmal rupture, as in our case where the patient presented with a perinephric haematoma secondary to renal artery bleeding. This uncommon but dangerous presentation reinforces the importance of prompt recognition of vasculitic features, particularly in patients presenting with unexplained systemic symptoms and organ involvement.
Vishal Kharb, Faizal Mohammed M, M.R.J. Qureshi, P. Garg, Vivek Saxena, Abhishek Rajendra Agarwal, Monica Mahajan. Polyarteritis Nodosa Masquerading as Fever. MMJ. 2025, June. Vol 2 (2).
References
- Handa R. Polyarteritis nodosa and microscopic polyangiitis – The Indian experience, Indian J Rheumatol. 2015;10(1S):S72-S77.
- Saghari S, Arslan K, Sordo S. Polyarteritis Nodosa With Complications: A Diagnostic Challenge and Management Dilemma. Cureus. 2023;15(11):e49677.
- Mukhopadhyay P, Rathi M, Kohli HS, et al. Polyarteritis nodosa presenting with spontaneous perirenal hematoma. Indian J Nephrol. 2012;22:295-7.
- Richard A Watts. Idiopathic polyarteritis nodosa – does it still exist? Viewpoint 1: as our knowledge makes progress, idiopathic polyarteritis nodosa is fading away. Rheumatology. 2025;64(Supplement_1):i79-81.
- Lightfoot RW Jr, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. 1990;33(8):1088-93.
- Stanton M, Tiwari V. Polyarteritis Nodosa. [Updated 2023 Feb 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482157/.
- Guillevin L, Mahr A, Cohen P, et al. French Vasculitis Study Group. Short-term corticosteroids then lamivudine and plasma exchanges to treat hepatitis B virus-related polyarteritis nodosa. Arthritis Rheum. 2004;51(3):482-7.
- Hernández-Rodríguez J, Hoffman GS. Updating single-organ vasculitis. Curr Opin Rheumatol. 2012;24(1):38-45.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49(7):750-6.
- Guillevin L, Pagnoux C. When should immunosuppressants be prescribed to treat systemic vasculitides? Intern Med. 2003;42(4):313-7.
- de Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med. 2009;150(10):670-80.
- Jayne D, Rasmussen N, Andrassy K, et al. European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349(1):36-44.
- Pagnoux C, Seror R, Henegar C, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum. 2010;62(2):616-26.