

Abstract:
Cerebral amyloid angiopathy (CAA) involves amyloid-beta accumulation in cortical and leptomeningeal vessel walls, increasing the risk of lobar haemorrhage. CAA pathogenesis likely involves impaired amyloid-beta clearance and prion-like spread. Specific genetic variants in amyloid precursor protein, apolipoprotein E, and other genes also modulate CAA risk. Clinical manifestations include lobar haemorrhages, cortical superficial siderosis, CAA-related inflammation, amyloid beta-related angiitis, cerebrovascular lesions, and sometimes dementia. MRI best detects CAA-related haemorrhages and microbleeds. Definitive diagnosis requires pathology, but modified Boston criteria allow categorisation as possible or probable CAA based on clinical and neuroimaging findings. Current management focuses on blood pressure control and cautious antithrombotic use to prevent haemorrhage recurrence. Experimental therapeutics target amyloid production, aggregation, clearance, inflammation, and neuroprotection, but clinical efficacy remains undetermined. Further research on CAA pathophysiology and rigorous clinical trials are warranted to elucidate genetic underpinnings, improve diagnostic accuracy, and develop effective treatments for this cerebral small vessel disease. The pathophysiology, genetics, clinical characteristics, diagnostics, and therapeutic methods of CAA are summarised in this study. The pathways connecting CAA to dementia, the role of neurovascular unit dysfunction, and the initiation of amyloid-beta vascular buildup are among the critical unanswered concerns. Improving our knowledge of CAA mechanisms and implementing newly developed therapies in clinical settings is essential to lessening the effects of this prevalent cerebrovascular illness
Key words: : Cerebral Amyloid Angiopathy, Amyloid-Beta, Lobar Haemorrhage, Neuroimaging, Clinical Trials
Introduction
Cerebral amyloid angiopathy (CAA) is a cerebrovascular disorder characterised by the deposition of amyloid-beta (Aβ) peptides, primarily Aβ40, in the walls of small to mediumsized arteries and arterioles of the cerebral cortex and leptomeninges.1,2 This vascular pathology leads to increased vessel fragility and an elevated risk of lobar intracerebral haemorrhages (ICHs), cortical superficial siderosis, and cognitive impairment.3
The pathogenesis of CAA is multifactorial and involves impaired clearance of Aβ peptides from the brain and a proposed "prion-like" spread of Aβ aggregates.4,5 The neurovascular unit, comprising neurons, glial cells, and vascular cells, is critical in maintaining cerebral blood flow and blood-brain barrier integrity. Dysfunction of this unit, particularly the pericytes, which regulate capillary blood flow, has been implicated in the development of CAA.6,7 Additionally, the "prion hypothesis" suggests that CAA progression may occur through the seeding and propagation of misfolded Aβ aggregates in a prion-like manner.8,9
Specific genetic variants in the amyloid precursor protein (APP), apolipoprotein E (APOE), and other genes have been identified as risk factors for CAA.10,11 Mutations in the APP gene, such as the Dutch E693Q mutation, have
been associated with severe CAA without the characteristic Alzheimer's disease neuropathology.12 The APOE ε4 allele is linked to an increased risk and severity of CAA, while the ε2 allele may increase the risk of CAA-related haemorrhages.13,14
The clinical manifestations of CAA are diverse and include lobar ICHs, cortical superficial siderosis, CAA-related inflammation, amyloid beta-related angiitis, cerebrovascular lesions, and in some cases, cognitive impairment.15,16 Accurate diagnosis of CAA relies on neuroimaging techniques, such as magnetic resonance imaging (MRI), which can detect CAA-related haemorrhages, micro-bleeds, and superficial siderosis.17 However, a definitive diagnosis often requires a pathological examination of brain tissue.18
Current management strategies for CAA focus on strict blood pressure control and careful evaluation of the risks and benefits of antithrombotic and anticoagulant therapies, as these agents may increase the risk of ICHs.19,20 Experimental therapeutic approaches targeting various aspects of the disease pathogenesis, including Aβ production, aggregation, clearance, inflammation, and neuroprotection, are under investigation.21-23 However, the clinical efficacy of these potential treatments remains to be determined.
In conclusion, CAA is a cerebrovascular disorder characterised by Aβ deposition in cortical and leptomeningeal vessels, leading to an increased risk of lobar haemorrhages and cognitive impairment. Further research into the pathophysiology of CAA, including neurovascular unit dysfunction and prion-like propagation roles and the development and evaluation of novel therapeutic strategies, is crucial for improving our understanding and management of this prevalent cerebrovascular disease.
Pathophysiology
In CAA, APP cleavage products deposit in vessel walls, predominantly Aβ40 and Aβ42.4,5 Unlike Alzheimer's dementia, where Aβ42 predominates, CAA features Aβ40 deposition.5,6 Aβ initially accumulates in the tunica media and adventitia, later spreading across all vessel layers, causing smooth muscle cell loss and vessel wall disruption.7
Impaired Aβ clearance likely contributes to CAA pathogenesis.8.9 The neurovascular unit, comprised of neurons, glia, and vascular cells, regulates cerebral blood flow and maintains blood-brain barrier integrity.10 Amyloid overexpression studies suggest neurovascular unit dysfunction in CAA.11 Pericytes play a crucial role in blood flow regulation, and recent evidence indicates Aβ40 exerts toxic effects on these cells.12,13
The "prion hypothesis" proposes CAA progression may occur via prion-like protein misfolding.14 Findings of seeding, spread of Aβ pathologies in animal models, and transmission of CAA pathology between humans support this idea.15-19 However, further substantiation is required regarding Aβ strains and conformational variations.20,21
Genetic Factors
Variants in APP, APOE, and other genes are linked to earlyonset CAA and haemorrhage.22-24 APP mutations at the Aβ region, like the E693Q Dutch mutation, often cause severe CAA without Alzheimer's neuropathology.25,26 Aβ40 and mutant Aβ42 are critical components of Dutch mutation vascular deposits.27 Mutations may increase Aβ aggregation, impede clearance, or enhance vascular affinity.28-31
APOE ε4 and ε2 alleles also modulate sporadic CAA risk. Meta-analysis confirms a dose-dependent ε4 association with CAA severity.32 ε2 may increase haemorrhage risk through vessel wall damage.33-35 Different mechanisms related to aggregation, clearance, and vessel wall responses likely underlie ε4 and ε2 contributions.36
Clinical Manifestations
Various clinical syndromes arise from CAA vasculopathy:37-42
- Lobar hemorrhages: Motor/sensory deficits, altered consciousness, visual loss, seizures
- Cortical superficial siderosis: Transient focal neurological episodes
- CAA-related inflammation: Subacute cognitive decline, headaches, focal deficits
- Amyloid beta-related angiitis: Rapid mental status decline, seizures, deficits
- Microbleeds/white matter lesions: Cognitive impairment, gait issues
- Convexity subarachnoid haemorrhage: Sudden neurological defects, headache
- Silent infarcts: Increased dementia and stroke risk
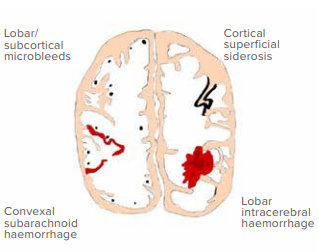
Figure 1: Showing various findings in the cerebral amyloid angiopathy (Hand drawn by Dr. Abhishek Dixit).
Diagnosis
MRI is the most effective imaging modality for detecting CAArelated microbleeds and haemorrhages.43 Digital subtraction angiography is not useful for visualising affected small vessels.44 A definitive diagnosis is only possible through post-mortem examination.45
The modified Boston criteria classify CAA as possible or probable based on clinical data, MRI findings, and exclusion of other causes.46 The Edinburgh computed tomography (CT)/genetic criteria aid diagnosis when MRI is unavailable.47 Clinical and radiological findings can support the diagnosis of inflammatory CAA syndromes, and some cases feature anti-amyloid beta antibodies in cerebrospinal fluid.48
| Category | Criteria |
|---|---|
| Definite CAA | 1. Full postmortem examination |
| 2. Lobar, cortical, or cortico-subcortical haemorrhage | |
| 3. Severe CAA with vasculopathy | |
| 4. Absence of another diagnostic lesion | |
| Probable CAA with supporting pathology | 1. Clinical data and pathological tissue (evacuated hematoma or cortical biopsy) |
| 2. Lobar, cortical, or cortico-subcortical haemorrhage | |
| 3. Some degree of CAA in the specimen | |
| 4. Absence of another diagnostic lesion | |
| Probable CAA | 1. Clinical data and MRI or CT |
| 2. Age ≥ 55 years | |
| 3. Presentation of spontaneous intracerebral haemorrhage | |
| 4. Presence of at least 2 of the strictly lobar haemorrhagic lesions on T2-weighted MRI (intracerebral haemorrhage, cerebral microbleeds, or foci of cortical superficial siderosis or convexity subarachnoid haemorrhage) OR one lobar haemorrhage with one of the white matter lesions (severe perivascular spaces in the central semiovale or white matter hyperintensities in a multisport pattern) | |
| 5. Absence of deep haemorrhagic lesions | |
| 6. Absence of other causes of haemorrhage or cortical superficial siderosis (cSS) | |
| Possible CAA | 1. Clinical data and MRI or CT |
| 2. Presentation with spontaneous intracerebral haemorrhage | |
| 3. Presence of one strictly lobar haemorrhage on T2-weighted MRI, cerebral microhaemorrhages, foci of cSS or convexity subarachnoid haemorrhage | |
| 4. Age ≥ 55 years | |
| 5. Absence of other causes of haemorrhage or cSS | |
| 6. One white matter lesion (severe perivascular spaces in the central semiovale or white matter hyperintensities in a multisport pattern) | |
| 7. Absence of deep haemorrhagic lesions | |
| 8. Absence of other causes of haemorrhages |
Table 1: Modified Boston Criteria Version 2.0 for the diagnosis of CAA
Abbreviations: CAA: cerebral amyloid angiopathy; MRI: magnetic resonance imaging; CT: computed tomography; cSS: Cortical superficial siderosis.
| Therapy | Mechanism | Stage of Trial |
|---|---|---|
| Cilostazol52 | Phosphodiesterase 3A inhibition promotes perivascular Aβ drainage | Preclinical |
| Taxifolin53 | Inhibits Aβ aggregation, antioxidant | Preclinical |
| Neprilysin up-regulation | Aβ-degrading enzyme reduces Aβ concentration | Preclinical |
| RAGE inhibition54 | Reduces Aβ accumulation in brain parenchyma | Preclinical |
| L-norvaline | Reduces BBB permeability, amyloid angiopathy, inflammation | Preclinical |
| Aβ12-28P peptide | Interferes with Aβ/APOE interaction | Preclinical |
| CPO-Aβ17-21P peptide | Interferes with Aβ/APOE interaction | Preclinical |
| Anti-APOE antibody55 | Modulates Aβ accumulation | Preclinical |
| Complement system targeting | Reduces chronic inflammatory response | Preclinical |
| Endothelial nitric oxide | Inhibits amyloidogenic processing of APP | Preclinical |
| Angiotensin receptor blockers56 | Induces Aβ reduction, neuroprotection, neurogenesis | Preclinical |
| Tau level modulation57 | Prevents neurodegeneration associated with CAA | Preclinical |
| BACE1 inhibitor58,59 | Decreases Aβ production | Preclinical |
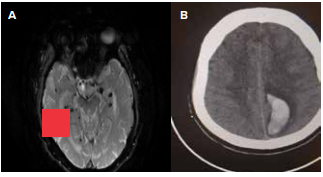
Figure 2: A) Susceptibility weighted imaging (SWi) in magnetic resonance imaging (MRI) showing multiple areas of blooming suggesting microhaemorrhages B) Computed tomography (CT) scan suggestive of lobar haemorrhage (Images courtesy Dr. Man Mohan Mehndiratta).
Treatment
Current management focuses on blood pressure control and cautious antithrombotic use to prevent incidents and recurrent haemorrhage (Table 1). Experimental therapeutics target Aβ production, aggregation, clearance, inflammation, and neuroprotection (Table 2). However, clinical efficacy remains to be determined. The current management of CAA focuses on strict blood pressure control and careful evaluation of the risks and benefits of antithrombotic and anticoagulant therapies.48,49 Maintaining a mean blood pressure above 65 mmHg is recommended to prevent incident and recurrent ICHs.48 However, using antithrombotic and anticoagulant agents requires carefully weighing the risks and benefits, as these therapies may increase the risk of ICHs in CAA patients.49
In addition to the standard management strategies, several experimental therapeutic approaches targeting various aspects of CAA pathogenesis are under investigation. One such agent is ponezumab (PF-04360365), an anti-Aβ40 selective antibody developed to prevent or reverse Aβ aggregation and deposition.50 Another candidate, NC-758 (cerebril), is an anti-Aβ agent being evaluated for treating lobar haemorrhage related to possible or probable CAA.51
Other experimental therapies aim to modulate different pathways involved in CAA, such as Aβ clearance, inflammation, and neurovascular unit dysfunction. For example, cilostazol, a phosphodiesterase 3A inhibitor, has shown promise in promoting perivascular Aβ drainage in preclinical studies.52 Taxifolin, an antioxidant compound, has been found to inhibit Aβ aggregation in preclinical models.53 Additionally, targeting the receptor for advanced glycation end products (RAGE) has been shown to reduce Aβ accumulation in the brain parenchyma,54 while APOE antibodies have the potential to modulate Aβ accumulation.55
Therapies targeting the renin-angiotensin system, such as angiotensin receptor blockers, have demonstrated Aβ reduction, neuroprotection, and neurogenesis in preclinical studies.56 Modulating tau levels has also been explored to prevent neurodegeneration associated with CAA.57 Lastly, inhibition of beta-secretase 1 (BACE1), an enzyme involved in Aβ production, has shown promise in reducing Aβ levels in preclinical models.58,59
While these experimental approaches hold promise, further rigorous clinical trials are necessary to determine their efficacy and safety in treating CAA. Continued research into the pathophysiology of CAA and developing novel therapeutic strategies are crucial for improving the management and outcomes of this prevalent cerebrovascular disease.
| Management/Treatment | Description | Stage/Results |
|---|---|---|
| Blood pressure control48 | Strict control of blood pressure to prevent incident and recurrent ICHs Mean above 65 mmHg |
Standard practice |
| Antithrombotics/anticoagulants49 | Careful evaluation and weighing of the risks and benefits of antithrombotics and anticoagulants | Standard practice |
| Ponezumab (PF-04360365)50 | Anti-Aβ40 selective antibody developed to prevent or reverse Aβ aggregation and deposition. | Phase 2 clinical trial completed. |
| NC-758 (cerebril)51 | Anti-Aβ agent to treat patients with lobar haemorrhage related to possible or probable CAA | Phase 2 clinical trial completed. |
Table 2: Current management and potential treatments for CAA.48-51
Abbreviations: ICHs: intracerebral haemorrhage; Aβ: amyloidbeta; CAA: cerebral amyloid angiopathy.
Table 3: Experimental therapeutic approaches for cerebral amyloid angiopathy (CAA).52-59
Abbreviations: Aβ: amyloid-beta; RAGE: receptor for advanced glycation end products; BBB: blood-brain barrier; APOE: apolipoprotein E; APP: amyloid precursor protein; CAA: cerebral amyloid angiopathy; BACE1: beta-secretase 1.
Please note that the information in this table is based on preclinical research and may not represent the complete list of experimental therapies. Further studies are required to determine the efficacy and safety of these approaches in treating CAA.
Conclusion
In summary, CAA is characterised by Aβ deposition in cortical and leptomeningeal vessels leading to lobar haemorrhages. Impaired Aβ clearance and prion-like spread may contribute to pathogenesis. Specific genetic variants also modulate CAA risk. Diagnosis relies on clinical presentation and neuroimaging findings. Therapeutic research is investigating multiple approaches to prevent vessel damage and haemorrhage. Further elucidation of CAA pathophysiology and clinical trials of emerging treatments are warranted.
Abhishek Dixit, Monalisa Vegda, Tanzeel Wani, Man Mohan Mehndiratta. Bridging the Knowledge Gap: The
Under-Reported Burden of Cerebral Amyloid Angiopathy. MMJ. 2025, March. Vol 1 (5).
References
- Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly.Annals of Neurology 2011;70(6):871-80.
- Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. Journal of Neurology, Neurosurgery & Psychiatry 2012;83(2):124-37.
- Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathologica. 2009;118:115-30.
- Attems J, Lintner F, Jellinger KA. Amyloid β peptide 1–42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathologica. 2004;107:283-91.
- Roher AE, Lowenson JD, Clarke S, et al. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proceedings of the National Academy of Sciences. 1993;90(22):10836-40.
- Keable A, Fenna K, Yuen HM, et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochimica et Biophysica Acta. 2016;1862(5):1037-46.
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. Journal of Neural Transmission.2002;109:813-36.
- Block F, Dafotakis M. Cerebral amyloid angiopathy in stroke medicine. Deutsches Ärzteblatt International. 2017;114(3):37.
- Jellinger K. Inverse relation between Braak stage and cerebrovascular pathology in Alzheimer predominant dementia. Journal of Neurology, Neurosurgery, and Psychiatry. 2000;68(6):799.
- Ikram MA, Wieberdink RG, Koudstaal PJ. International epidemiology of intracerebral hemorrhage. Current atherosclerosis reports. 2012;14:300-6.
- Giroud M, Gras P, Chadan N, et al. Cerebral haemorrhage in a French prospective population study. Journal of Neurology, Neurosurgery & Psychiatry. 1991;54(7):595-8.
- Bateman BT, Claassen J, Willey JZ, et al. Convulsive status epilepticus after ischemic stroke and intracerebral hemorrhage: frequency, predictors, and impact on outcome in a large administrative dataset. Neurocritical care. 2007;7:187-93.
- Yamada M. Risk factors for cerebral amyloid angiopathy in the elderly. Annals of the New York Academy of Sciences. 2002;977(1):37-44.
- Yamada M. Cerebral amyloid angiopathy: emerging concepts. Journal of stroke. 2015;17(1):17.
- Huynh TP, Davis AA, Ulrich JD, et al. Apolipoprotein E and Alzheimer's disease: the influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins: Thematic Review Series: ApoE and Lipid Homeostasis in Alzheimer's Disease. Journal of lipid research. 2017;58(5):824-36.
- Nicoll JA, Burnett C, Love S, et al. High frequency of apolipoprotein E ϵ2 allele in hemorrhage due to cerebral amyloid angiopathy. Annals of neurology. 1997;41(6):716-21.
- Yamada M, Sodeyama N, Itoh Y, et al. Association of presenilin-1 polymorphism with cerebral amyloid angiopathy in the elderly. Stroke. 1997;28(11):2219-21.
- Yamada M. Cerebral amyloid angiopathy and gene polymorphisms. Journal of the neurological sciences. 2004;226(1-2):41-4.
- Yamada M, Sodeyama N, Itoh Y, et al. Association of α1‐antichymotrypsin polymorphism with cerebral amyloid angiopathy. Annals of Neurology. 1998;44(1):129-31.
- Charidimou A, Jaunmuktane Z, Baron JC, et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology. 2014;82(1):57-62.
- Rensink AA, de Waal RM, Kremer B, et al. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207-23.
- Smith EE. Cerebral amyloid angiopathy as a cause of neurodegeneration. Journal of neurochemistry. 2018;144(5):651-8.
- Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96(1):17-42.
- Niwa K, Younkin L, Ebeling C, et al. Aβ1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proceedings of the National Academy of Sciences. 2000;97(17):9735-40.
- Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55-60.
- Schultz N, Brännström K, Byman E, et al. Amyloid‐beta 1‐40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell. 2018;17(3):e12728.
- Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. Nature Reviews Immunology. 2013;13(12):888-902.
- Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nature Reviews Genetics. 2018;19(7):405-18.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781-4.
- Di Fede G, Catania M, Maderna E, et al. Molecular subtypes of Alzheimer’s disease. Scientific reports. 2018;8(1):3269.
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Annals of neurology. 2011;70(4):532-40.
- Burwinkel M, Lutzenberger M, Heppner FL, et al. Intravenous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta neuropathologica communications. 2018 Dec;6:1-6.
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature reviews Molecular cell biology. 2010;11(4):301-7.
- Cali I, Cohen ML, Haïk S, et al. Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: an international study. Acta neuropathologica communications. 2018;6:1-9.
- Purro SA, Farrow MA, Linehan J, et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 2018;564(7736):415-9.
- Banerjee G, Adams ME, Jaunmuktane Z, et al. Early onset cerebral amyloid angiopathy following childhood exposure to cadaveric dura. Annals of neurology. 2019;85(2):284-90.
- Jaunmuktane Z, Quaegebeur A, Taipa R, et al. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta neuropathologica. 2018;135:671-9.
- Giaccone G, Maderna E, Marucci G, et al. Iatrogenic early onset cerebral amyloid angiopathy 30 years after cerebral trauma with neurosurgery: vascular amyloid deposits are made up of both Aβ40 and Aβ42. Acta neuropathologica communications. 2019;7:1-4.
- Hervé D, Porché M, Cabrejo L, et al. Fatal Aβ cerebral amyloid angiopathy 4 decades after a dural graft at the age of 2 years. Acta neuropathologica. 2018;135:801-3.
- Benbadis SR. Editorialist response: Adherence with psychotherapy and treatment outcomes for psychogenic nonepileptic seizures. Neurology. 2019;93(22):981-2.
- Caldas AC, Silva C, Albuquerque L, et al. Cerebral amyloid angiopathy associated with inflammation: report of 3 cases and systematic review. Journal of Stroke and Cerebrovascular Diseases. 2015;24(9):2039-48.
- Nandigam RN, Viswanathan A, Delgado P, et al. MR imaging detection of cerebral microbleeds: effect of susceptibility-weighted imaging, section thickness, and field strength. American Journal of Neuroradiology. 2009;30(2):338-43.
- Haacke EM, DelProposto ZS, Chaturvedi S, et al. Imaging cerebral amyloid angiopathy with susceptibility-weighted imaging. American journal of neuroradiology. 2007;28(2):316-7.
- Charidimou A, Boulouis G, Frosch MP, et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI-neuropathology diagnostic accuracy study. Lancet Neurol. 2022, 29:1474-4422.
- Rodrigues MA, Samarasekera N, Lerpiniere C, et al. The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: model development and diagnostic test accuracy study. The Lancet Neurology. 2018;17(3):232-40.
- Weber SA, Patel RK, Lutsep HL. Cerebral amyloid angiopathy: diagnosis and potential therapies. Expert Review of Neurotherapeutics. 2018;18(6):503-13.
- Punetha J, Karaca E, Gezdirici A, et al. Biallelic CACNA2D2 variants in epileptic encephalopathy and cerebellar atrophy. Annals of Clinical and Translational Neurology. 2019;6(8):1395-406.
- Maki T, Okamoto Y, Carare RO, et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β‐amyloid. Annals of clinical and translational neurology. 2014;1(8):519-33.
- Maki T, Okamoto Y, Carare RO, et al. Phosphodiesterase III inhibitor promotes drainage of cerebrovascular β‐amyloid. Annals of clinical and translational neurology. 2014;1(8):519-33.
- Kimura T, Hamazaki TS, Sugaya M, et al. Cilostazol improves lymphatic function by inducing proliferation and stabilization of lymphatic endothelial cells. Journal of Dermatological Science. 2014;74(2):150-8.
- Sumbria RK, Vasilevko V, Grigoryan MM, et al. Effects of phosphodiesterase 3A modulation on murine cerebral microhemorrhages. Journal of neuroinflammation. 2017;14:1-4.
- Saito S, Yamamoto Y, Maki T, et al. Taxifolin inhibits amyloid-β oligomer formation and fully restores vascular integrity and memory in cerebral amyloid angiopathy. Acta neuropathologica communications. 2017;5:1-6.
- Inoue T, Yamakage H, Tanaka M, et al. Oxytocin suppresses inflammatory responses induced by lipopolysaccharide through inhibition of the eIF-2α–ATF4 pathway in mouse microglia. Cells. 2019;8(6):527.
- Iwata N, Mizukami H, Shirotani K, et al. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. Journal of Neuroscience. 2004;24(4):991-8.
- Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-β 1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. The Journal of clinical investigation. 2000;106(12):1489-99.
- Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine. 2003;9(7):907-13.
- Deane R, Singh I, Sagare AP, et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. The Journal of clinical investigation. 2012;122(4):1377-92.