

Abstract:
Inflammatory demyelinating polyradiculoneuropathies are acquired immune mediated polyneuropathies. The most common variants are acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor sensory axonal neuropathy (AMSAN), and Miller Fisher syndrome (MFS). Electro-diagnostic studies are the mainstay for diagnosis and early management for subtypes and variants. Multifocal demyelination is the hallmark of AIDP. In the first week of symptoms, increase in cerebrospinal fluid (CSF) protein and immunoglobulins is not associated with a cellular response (albumino-cytological dissociation). Elevated anti-GQ1b anti-ganglioside IgG antibodies are the hallmark of 95%-98% of MFS. High titres of anti-GM1 (ganglioside-monosialic acid), anti-GD1b, anti-GD1a IgG anti-gangliosides antibodies have been associated with the AMAN variant. Differential diagnosis includes acute intermittent porphyria, acute transverse myelitis, hypokalaemic periodic paralysis, etc. It is organ specific immune mediated disorder caused by synergistic cell mediated and humoral immune responses against peripheral nerve antigen. Two thirds of the patients have a preceding event. The management includes general supportive treatment, human immunoglobulins also known as intravenous immunoglobulin (IVIg) and plasma exchange (PLEX). The prognosis depends on the age of the patient, onset of symptoms, disease progression, type of variants and earlier treatment.
Key words: Guillain-Barre Syndrome (GBS), Inflammatory Demyelinating Polyradiculoneuropathies, Human Immunoglobulin or Intravenous Immunoglobulin (IVIg).
Introduction
Inflammatory demyelinating polyradiculoneuropathies are considered acquired immune-mediated polyneuropathies, meaning they are a group of nerve disorders caused by an abnormal immune response that develops over time, attacking the myelin sheath surrounding peripheral nerves. In 1916, Guillain-Barre syndrome (GBS) was coined by Guillain, Barre and Strohl, who focused on the main clinical features of- motor weakness, areflexia, paraesthesia with minor sensory loss and albumino-cytological dissociation (increased protein in cerebrospinal fluid [CSF] without pleocytosis). The diagnosis criteria was initially1 based on clinical criteria (progressive weakness of limbs, areflexia or hyporeflexia); supportive criteria (progressive over 4 weeks, symmetry of symptoms and signs, bifacial palsies, autonomic dysfunction, absence of fever at onset, recovery beginning 4 weeks after progression ceases); elevated CSF protein with <10 cells/microL; electrodiagnostic features of nerve conduction slowing or block.
Classification
The classification of GBS subtypes and variants (Table 1) depends on preceding infection, diminished reflexes, and elevated CSF protein levels (immune-mediated origin).2-11
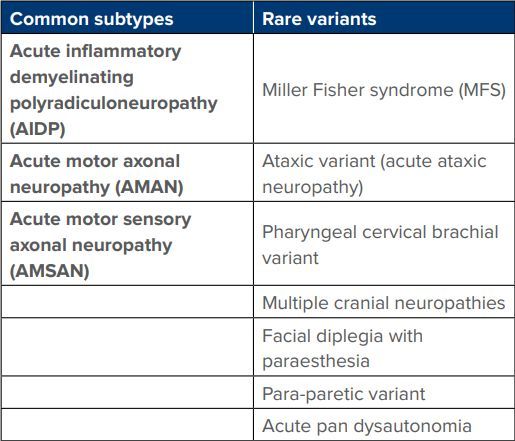
Table 1: Classification of Guillain-Barre syndrome (GBS) subtypes and variants.
Acute inflammatory demyelinating polyradiculoneuropathy (AIDP)
The diagnosis is based on clinical criteria, antecedent events, and electrodiagnostic criteria (Brighton criteria). It is the classic form of GBS, nonseasonal, affects all ages, and male to female ratio is 1.5:1. It is the leading cause of acute paralytic disease in western countries with mean annual incidence of 1.8/100000 population.2 The patients present with classical symmetrical weakness of lower limbs ascending upwards and proximally presents over hours to days, and involves arms, facial, oropharyngeal muscles, and in severe cases respiratory muscles with range from mild symptoms to quadriplegia. Hyporeflexia or areflexia is one of the cardinal features. Cranial nerve involvement is seen in 45%-75% cases with facial paresis followed by extraocular and lower cranial nerves involvement. Respiratory failure and ventilation are required in extremes of age and in 9%-30% cases.3 Sensory involvement in the form of impaired distal vibration sense, moderate to severe pain in the extremities, interscapular area or back in 70% cases in acute phase of illness. Autonomic dysfunction noted in 65% cases in 2-4 weeks of illness. The symptoms and signs include orthostatic hypotension, urinary retention, gastrointestinal atony, iridoplegia, hypertension, sinus tachycardia, tachyarrhythmias, anhidrosis, diaphoresis, vagal spells triggered by tracheal suctioning; most commonly in severe quadriplegia and respiratory failure (exclude pulmonary embolism; hypoxia; pneumonia).
Acute motor axonal neuropathy (AMAN)
It's a subtype of GBS that was first identified in China during an epidemic affecting children and young adults, and it has since become the most common subtype in Asia. It presents with acute flaccid paralysis with no sensory involvement. It differs from AIDP with normal SNAPs (sensory nerve action potentials) and reduced CMAP (compound muscle action potentials) amplitude with no demyelination. Most patients improve as rapidly as AIDP. Nerve biopsy revealed conduction failure of motor axons with macrophage intrusion or axonal degeneration of motor nerve terminals. Majority of the patients (76%) had antecedent Campylobacter jejuni infection (molecular mimicry), and mainly anti-GM1 or anti-GD1a antibodies (ganglioside-like epitopes) lead to membrane attack complex formation and conduction failure.4
Acute motor sensory axonal neuropathy (AMSAN)
It is a fulminant and less common subtype of GBS with poor prognosis of recovery. Acute and rapidly progressive quadriparesis and with muscle wasting and prolonged ventilatory support. It is different from classic GBS with markedly reduced or absent CMAPs and absent SNAPs without significant conduction slowing in electro diagnostics study. Primary axonopathy is the underlying cause with extensive axonal degeneration without significant inflammation or demyelination in ventral and dorsal roots and peripheral nerves.5
Miller Fisher syndrome (MFS)
It is a rare subtype with 5% of cases with triad of ophthalmoplegia, ataxia, and areflexia. The patients present with diplopia (complete ophthalmoplegia, dilated, and unreactive pupils to external ophthalmoparesis with or without ptosis) with or without other cranial nerves followed by gait and limb ataxia. Motor strength is characteristically preserved. It is different from classic GBS (AIDP) with normal electrodiagnostic study except sensory nerve of upper limbs involvement and relatively preserved sural SNAPs. IgG antibodies against ganglioside GQ1b is related to the pathogenesis and seen in 98% of active disease. CSF shows albumin cytological dissociation 1 week after the onset. Magnetic resonance imaging (MRI) of brain is normal or shows brainstem abnormality or cranial nerve enhancement. The patient may have hyperreflexia instead of areflexia (Bickerstaff Fisher syndrome, a variant of the anti-GQ1b syndrome within the MFS spectrum). MFS has a favourable prognosis with recovery in 10 weeks and reduction in antibody titres.6,7
Acute pan dysautonomia
It is a rare subtype of GBS with rapid onset of severe orthostatic hypotension, heat intolerance, anhidrosis, dry eyes, dry mouth, fixed pupils, fixed heart rate, bladder and bowel disturbances. The deep tendon reflexes are usually absent with preserved motor and sensory system. Anti-ganglionic receptor blocking antibodies are elevated in all autoimmune autonomic neuropathies.8
Pharyngeal-cervical-brachial variant
Isolated facial diplegia with distal paresthesia is the cardinal feature of this rare variant. It is a strictly regional variant with rapidly progressive multiple cranial neuropathies (polyneuritis cranialis).9
Sensory ataxic variant
It is a very rare variant with acute ataxia, sensory loss, areflexia, high CSF proteins and demyelination in electro diagnostics. SNAPs are intact in 60% cases. It is different from MFS with absent ophthalmoplegia; only 65% have elevated anti GQ1b antibodies.10
Acute painful small fiber neuropathy
It is a rare variant and is cortico steroid-responsive, unlike other variants. Antecedent cytomegalovirus (CMV) infection results in sensory predominant GBS.11
Diagnosis
Electrodiagnostic studies are the mainstay for diagnosing subtypes and variants, as well as for guiding early management. Multifocal demyelination is the hallmark of AIDP. It confirms the diagnosis of GBS. Majority of the cases (90%) have some form of abnormalities of distal motor latencies and F wave latencies with 30 days onset, 30% cases with partial motor conduction block and 24% of cases have motor conduction velocity.12
Cerebrospinal fluid (CSF) analysis
In the first week of symptoms, increase in CSF protein and immunoglobulins is not associated with a cellular response (albumino-cytological dissociation).10,12 In 10% of cases, CSF protein levels may be normal; in 10% of cases, there may be >10 cells/mm; and in cases associated with HIV or Lyme disease, CSF may contain >50 cells/mm.10,12
Serological tests
No specific serological tests for AIDP. Elevated anti-GQ1b anti ganglioside IgG antibodies are the hallmark of 95%-98% of MFS. High titres of anti-GM1, anti-GD1b, anti-GD1a IgG anti gangliosides antibodies have been associated with AMAN variant.13
Imaging studies
MRI of the brain and spine is most useful for excluding brainstem or spinal cord disease, which is typically normal in these cases. Rarely, MRI brain may show brain stem abnormalities or cranial nerve enhancement. MRI spine gadolinium may show lumbar roots and plexus enhancement in children with GBS.14
Treatment
The mainstay of treatment of GBS is general supportive management.
General supportive treatment
Rapidly worsening acute GBS should be observed in intensive care unit (ICU) care with prevention of complications like respiratory failure and autonomic dysfunction. Ventilatory care is needed in patients with rapid disease progression (onset less than 7 days); severity of limbs weakness; facial weakness; bulbar weakness, 20-30-40 rule is followed for intubation and ICU care.10,14 Continuous ECG and blood pressure monitoring for cardiac arrhythmias or dysautonomia.
- Use of anti-hypertensive drugs with caution
- Careful tracheal suctioning to avoid sudden episodes of hypotension or bradyarrhythmia
- Use of NSAIDS (nonsteroidal anti-inflammatory drugs) for back and radicular pains
- Early enteral feedings with high calorie protein diet
- Low molecular weight heparin subcutaneously with calf compression devices
- Prevention and prompt treatment of nosocomial infections of urine and chest
- Chest physiotherapy and frequent oral suctioning. Tracheostomy if ventilatory care is required for more than 2 weeks
- Skilled nursing care, including the management of the eyes, skin, mouth, bowel, and bladder, is essential
- Avoid exposure keratitis; avoid pressure induced ulnar or fibular palsies
- Physiotherapy to be started early to prevent contractures, joint immobilisation, and venous stasis; proper positioning and padding; side turning to pressure sores. In recovery phase; skilled physiotherapy, occupational therapy and rehabilitation should be performed11
Specific interventions
Human immunoglobulins (IVIg) and plasma exchanges (PLEX) have been shown to be equally effective.10
Plasma exchange (PLEX
Six large, randomised controlled trials including more than 600 patients have established the benefit of PLEX in acute GBS (within 14 days) by shortening the recovery time.12 Cochrane review showed PLEX to be superior over supportive care in hastening the recovery within 30 days of onset.13 Therapeutic PLEX are done for moderate to severe weakness within 2 weeks of onset (ability to walk with support or worse). In mild disease, 2 sittings of plasma exchange are enough for recovery. 4 exchanges were equally beneficial as compared to 6 exchanges in moderate to severe disease (French cooperative group on plasma exchange, 1997).13 Schedule: four to five exchanges (40-50 mL/kg of body weight) with a continuous flow machine on alternate days using saline and albumin as replacement fluids. It should be performed in centres with experience in exchange techniques in critically ill patients. Complications include haematoma, pneumothorax, and catheter induced sepsis. It is contraindicated in septicaemia, active bleeding, and severe cardiovascular instability.13
Human immunoglobulins (IVIg)
Three randomised control trials and a Cochrane systemic review comparing IVIg with PLEX showed the benefit of high dose IVIg and were equally effective in both adults and children with no advantage of using both together. The dosage used was 5 daily infusions of IVIg (0.4 gm/kg body weight per day) given within 2 weeks of the disease onset.10,11,12,14 Mode of action: anti idiotypical antibodies in IVIg bind and neutralise pathogenetic antibodies.14 Various adverse effects of human immunoglobulins has been listed in Table 2.
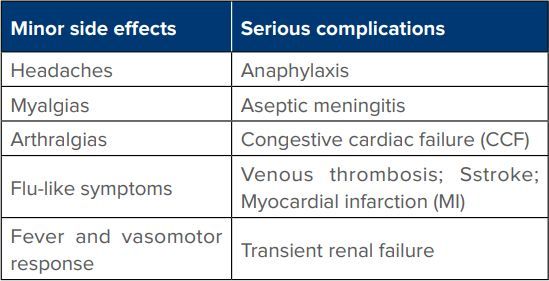
Table 2: Side effects of human immunoglobulins.
The side effects can be avoided by slow intravenous infusion and by giving premedication with oral paracetamol 1 gm; or ibuprofen 800 mg few hours before each infusion and repeat 6 hours later if headache develops.10,14 Contraindications: hyper viscosity; congestive heart failure; chronic renal failure (diabetes associated); congenital Ig A deficiency.14,15 In such cases plasma exchange is preferred.
Treatment related fluctuations is seen in 10%-20% patients with both IVIg and plasma exchange. Need to differentiate from rapid onset of CIDP (chronic inflammatory demyelinating polyneuropathy). Rescue IVIg and plasma exchanges in poorly responsive or relapsing GBS may be useful. Monitoring of IgG levels may be helpful in poorly responsive cases.15
Corticosteroids are not recommended as two randomised control trials found no benefit of alone or combined with IVIg as compared to IVIg alone in hastening recovery. A Cochrane review confirmed that it does not have significant benefit or harm.16
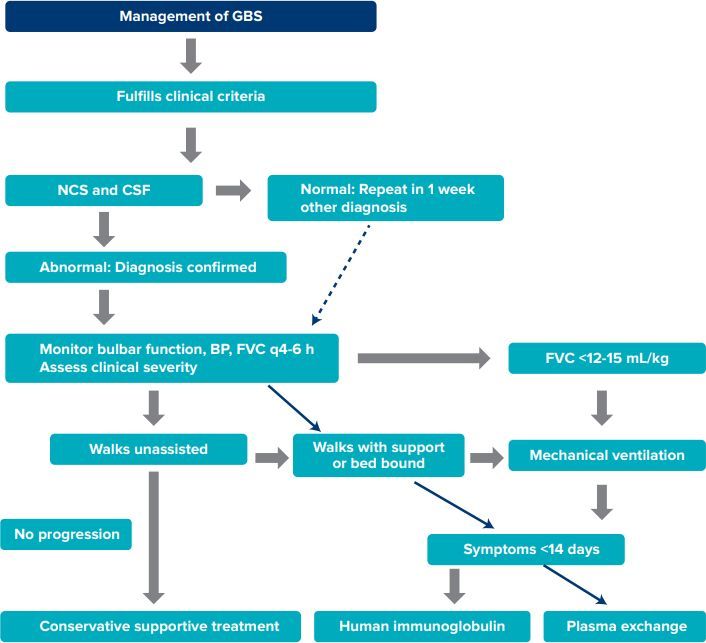
Flow chart 1: Management of Guillain-Barre syndrome (GBS) flow chart.16
Abbreviations: GBS: Guillain-Barre syndrome; NCS: nerve conduction studies; CSF: cerebrospinal fluid; BP: blood pressure; FVC: forced vital capacity.
Course and Prognosis
Fifteen percent of patients experience a mild condition with recovery within a few weeks, without long-term walking disability. In 5%-20% of cases, the disease takes a fulminant course, leading to flaccid quadriplegia, ventilator dependence, and axonal degeneration within 2 days of onset. Recovery is delayed, and patients may have incomplete or significant residual motor deficits at 1-year follow-up. AMAN and AIDP share a similar prognosis. Around 30% of patients develop respiratory insufficiency requiring assisted ventilation, and 2%-5% die due to complications.11
Conclusion
It is very important to diagnose GBS and its variants as early as possible to ensure proper management and avoid complications and poor long-term prognosis. IVIg have revolutionised the management and early recovery of most of the patients with less disability and good prognosis. GBS and its variants overall carry a good prognosis in recovery with supportive care; IVIg and plasma exchange in good neurology intensive care units all over the world with low mortality.
Manoj Khanal. Guillain-Barre Syndrome and Its Variants. MMJ. 2025, March. Vol 1 (5).
References
- Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27 Suppl:S21-4.
- Sejvar JJ, Kohl KS, Gidudu J, et al. Guillain-Barré syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011;29(3):599.
- Alshekhlee A, Hussain Z, Sultan B, et al. Guillain-Barré syndrome: incidence and mortality rates in US hospitals. Neurology. 2008;70(18):1608-13.
- Ho TW, Li CY, Cornblath DR, et al. Patterns of recovery in the Guillain-Barre syndromes. Neurology. 1997;48(3):695-700.
- Koga M, Kishi M, Fukusako T, et al. Antecedent infections in Fisher syndrome: sources of variation in clinical characteristics. Journal of Neurology. 2019;266:1655-62.
- Vernino S, Wolfe GI. Antibody testing in peripheral neuropathies. Neurologic Clinics. 2007;25(1):29-46.
- Susuki K, Koga M, Hirata K, et al. A Guillain-Barré syndrome variant with prominent facial diplegia. Journal of Neurology. 2009;256:1899-905.
- Oh SJ, LaGanke C, Claussen GC. Sensory Guillain–Barré syndrome. Neurology. 2001;56(1):82-6.
- Dabby R, Gilad R, Sadeh M, et al. Acute steroid responsive small-fiber sensory neuropathy: a new entity?. Journal of the Peripheral Nervous System. 2006;11(1):47-52.
- Leonhard SE, Mandarakas MR, Gondim FA, et al. Diagnosis and management of Guillain–Barré syndrome in ten steps. Nature Reviews Neurology. 2019;15(11):671-83.
- Neurology in clinical practice. 8th edition. Volume two. Jankovic et al. 2022. 1884.
- Raphaël JC, Chevret S, Hughes RA, et al. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2012;(7):CD001798.
- Appropriate number of plasma exchanges in Guillain-Barré syndrome. The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Ann Neurol. 1997;41(3):298-306.
- Hughes RA, Swan AV, Raphaël JC, et al. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130(9):2245-57.
- Alboudi AM, Sarathchandran P, Geblawi SS, et al. Rescue treatment in patients with poorly responsive Guillain–Barre syndrome. Sage Open Medicine. 2019;7:2050312119840195.
- Bosch EP. Guillain-Barre syndrome: an update of acute immune-mediated polyradiculoneuropathies. The Neurologist. 1998;4(4):211-26.